




















Зміст:
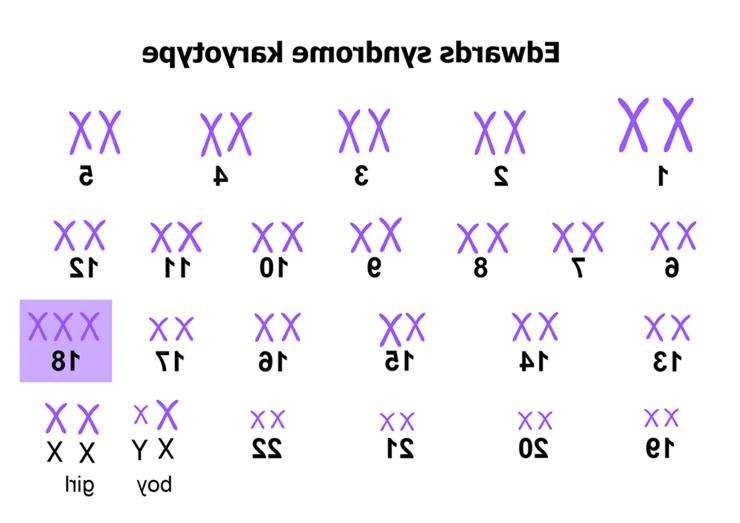
Синдром Едвардса відносять до хромосомних патологій. Він характеризується появою додаткової хромосоми у 18-ій парі. При розвитку захворювання у хворого відзначаються особливості зовнішнього вигляду: зменшення розмірів черепа, очей і зміна форми вух. Порушується будова внутрішніх органів — серця, судинної системи, органів сечовидільної системи та ін. Виявити трисомії можна внутрішньоутробно. Для цього існують інвазивні та неінвазивні методи дослідження. Ефективне лікування не розроблено. Можлива тільки симптоматична терапія, знижує вираженість клінічних ознак.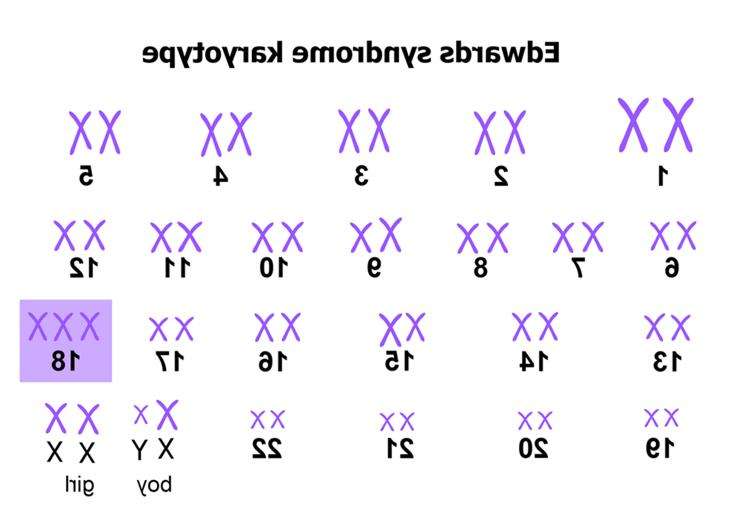 Синдром Едвардса характеризується трьома 18 хромосомами
Синдром Едвардса характеризується трьома 18 хромосомами
Зміст
Причини розвитку
Основна причина розвитку захворювання — зміна хромосомного набору. Трисомія 18 зустрічається найчастіше. У деяких випадках у пацієнтів виявляють мозаїчний варіант патології, що супроводжується мутаціями в певних генах.
Збільшення кількості 18 хромосоми виникає в результаті її нерозходження між клітинами при мейозі (процес поділу статевих клітин). Порушення частіше виникають у яйцеклітинах. У дитини розвивається важка форма захворювання. Легкі варіанти хвороби пов’язані з мозаїцизмом. Це ситуація, при якій частина хромосомного матеріалу переноситься, приводячи до порушення роботи генів.
У розвитку хромосомних патологій важливих факторів є вік жінки. Якщо вагітність виникла після 30 років, то ризик виникнення трисомії збільшується в кілька разів.
Клінічні прояви
Перші зміни при синдромі Едвардса відзначаються в період вагітності. При обстеженні жінки виявляють багатоводдя, зменшення розмірів плаценти і наявність тільки однієї пупкової артерії (в нормі – 2 артерії). Маса дітей при народженні менше норми – до 2100 м і менше, що пов’язано із загальною гіпотрофією.
У дітей з патологією виражені фенотипічні зміни (особливості зовнішнього вигляду). При їх наявності лікар проводить додаткове обстеження для виключення синдрому Патау та інших хромосомних аномалій. До характерних симптомів відносять:
- долихоцефалические зміни форми черепа. Поздовжній розмір голови переважає над поперечним;
- зменшення розміру лоба при виступаючому потилиці;
- у дітей зменшені очі і рот.
Рідше зустрічається шкірна складка біля внутрішнього краю ока, опущення нижнього століття, екзофтальм (“вирячені очі), косоокість та укорочення шиї. Деформація вух представлена зменшенням мочок, відсутністю козелків і низьким розташуванням вушних раковин.
Ознаки захворювання включають зміни опорно-рухового апарату: деформацію пальців кистей, зменшення розмірів грудини і ребер, вроджений вивих стегна, клишоногість та ін. У зв’язку з порушенням розвитку тканин у хворих з’являються папіломи і гемангіоми на шкірному покриві.
18 хромосома у людини відіграє важливу роль у формуванні внутрішніх органів. Збільшення кількості її копій або транслокація (зміна розташування) ділянок призводить до аномалій їх розвитку. Найбільш часто зустрічаються дефекти серцево-судинної системи у вигляді коарктації аорти, тетради Фалло, зміни положення клапанів та ін. Також страждає шлунково-кишковий тракт, сечостатева система і центральна нервова система. Зміни в будові головного мозку характеризуються гідроцефалією, кістами, гіпоплазією окремих структур і т. п.
Синдром Едвардса призводить до загибелі пацієнтів в перший рік життя. Смерть виникає при відсутності лікування через розвиток дихальної або серцево-судинної недостатності.
Негативні наслідки
Порушення розвитку внутрішніх органів та центральної нервової системи призводить до множинних вад розвитку. У зв’язку з аномаліями будови серця, головних судин і органів дихання у дитини розвивається серцево-судинна чи дихальна недостатність. Вона може стати причиною смерті при відсутності своєчасної медичної допомоги.
Вади розвитку органів сечовиділення створюють передумову для виникнення пієлонефриту, поликистозной нирки та інших аномалій. В результаті їх прогресування розвивається хронічна ниркова недостатність, що супроводжується накопиченням в крові продуктів азотистого обміну. Це може призвести до порушення роботи ЦНС та інших наслідків, навіть до смерті.
Наслідками мікроцефалії та гіпоплазії мозкових структур є функціональні розлади роботи внутрішніх органів, а також неможливість формування складних психічних або рухових навичок. При ураженні центрів, відповідальних за дихання або серцево-судинну діяльність, розвивається їх недостатність.
Діагностика хвороби
В діагностиці захворювання важливо її внутрішньоутробний виявлення. При виявленні хвороби показано штучне переривання вагітності. Непряма ознака патології — недорозвинення однією з пупкових артерій, багатоводдя, зменшення розмірів плаценти та ін. Вони можуть бути виявлені під час проведення планового УЗД.
Максимальної діагностичною цінністю володіють неінвазивні методи, пов’язані з визначенням у крові матері маркерів хвороби: хоріонічного гонадотропіну, PAPP, альфа-фетопротеїну, а також вільного естрадіолу. Зміна рівня зазначених речовин у крові жінки є показанням для проведення додаткових діагностичних процедур.
У процесі діагностики захворювання велике значення має сукупність факторів: результати лабораторного скринінгу і даних УЗД, термін гестації і вік вагітної жінки. При наявності декількох факторів рекомендовані інвазивні дослідження — біопсія хоріона, амніоцентез (взяття на аналіз навколоплідних вод) або кордоцентез (взяття аналізу крові з пуповини). Отриманий в результаті процедур матеріал плода піддається кариотипированию з підрахунком числа хромосом.
Хворі після встановлення діагнозу потребують комплексному обстеженні. Рекомендується провести консультації невролога, кардіохірурга, кардіолога, уролога та інших лікарів. Всім дітям проводять електрокардіографію, Ехокг та ультразвукове дослідження внутрішніх органів. При наявності неврологічного дефіциту необхідно зняття ЕЕГ і УЗД або КТ головного мозку.
Диференціальну діагностику проводять з різними хромосомними патологіями – синдром котячого крику і пр. В її основі лежать молекулярно-генетичні методи та визначення каріотипу.
Підходи до лікування
 Синдром Едвардса видно на УЗД і вимагає комплексного лікування
Синдром Едвардса видно на УЗД і вимагає комплексного лікування
Хромосомні аномалії не можуть бути усунені. У зв’язку з цим проведене лікування спрямоване на підтримання життя хворого і усунення наявних симптомів. Терапія включає медикаментозні, немедикаментозні та хірургічні методики.
Лікарські препарати призначаються в таких випадках:
- наявність інфекційного ураження органів сечовидільної або дихальної системи. Застосовують антибіотики широкого спектру дії, переважно з групи захищених пеніцилінів або цефалоспоринів;
- препарати, що поліпшують мозковий кровообіг;
- засоби, що регулюють тонус кровоносних судин.
Важливий аспект лікування — регулярні заняття лікувальною фізкультурою і сеанси масажу. ЛФК необхідна для нормалізації м’язового тонусу, зняття спазмів м’язів. Фізичні вправи покращують моторику хворого. Діти повинні постійно перебувати під медичним наглядом для своєчасного виявлення ускладнень та надання допомоги.
Хірургічні втручання виконуються з приводу аномалій розвитку серця, судинного русла та органів сечовидільної системи. Операції спрямовані на стабілізацію їх стану і поліпшення якості життя дитини.
Прогноз для дитини
Прогноз при хромосомних захворюваннях несприятливий. Одужання неможливе. Середня тривалість життя — 1-2 роки. До віку 12 місяців доживає менше 10% хворих, а до 10 років — лише 1%. Для мозаїчних варіантів синдрому Едвардса прогноз сприятливіші, що пов’язано з меншою вираженості аномалій внутрішніх органів.
Основні причини загибелі пов’язані з декомпенсацією серцево-судинної і дихальної недостатності. Летальні стану розвиваються раптово і характеризується прогресивним перебігом, незважаючи на проведену терапію.
Можливості профілактики
Профілактика хвороби грунтується на усуненні причин, які підвищують ризик розвитку хромосомних аномалій. До них відносять:
- вагітність у віці до 30 років. Чим вік батьків вище, тим ризик виникнення змін у хромосомах більше;
- уникати роботи на шкідливому хімічному та промисловому виробництві;
- виключення шкідливих звичок — тютюнопаління, вживання алкоголю та наркоманії. Зазначені впливу у період вагітності та після нього сприяють накопиченню в статевих клітинах мутацій, що може привести до безлічі хромосомних аномалій у майбутнього малюка, змінити його каріотип.
Важливим елементом профілактики захворювання є скринінг вагітних. Зміни, характерні для синдрому, добре видно на УЗД. Призначення додаткових інвазивних методик дозволяє підтвердити діагноз і провести медикаментозний аборт.
Розвиток синдрому Едвардса пов’язано із змінами в хромосомному наборі плода. Захворювання характеризується несприятливим прогнозом, так як більшість дітей вмирає в перші роки життя на тлі серцево-судинної або дихальної недостатності. Лікування патології носить симптоматичний характер, так як виправлення набору хромосом неможливо. Терапія спрямована на усунення конкретних симптомів та збільшення тривалості життя хворої дитини. В процесі обстеження важливо виключити інші хромосомні аномалії — синдром Шерешевського-Тернера та ін Це дозволяє уточнити діагноз і прогноз для хворого.
Читайте в наступній статті: синдром денді уокера







{kind=link}